

By Erick B. Iezzi, Eugene Camerino, Grant C. Daniels, and James H. Wynne, U.S. Naval Research Laboratory
Crosslinked networks, such as two-component polyurethanes, are used for numerous commercial applications because of their unique mechanical, thermal, and hydrocarbon resistant properties. However, the covalently bonded linkages in these materials are irreversible, which renders them difficult to degrade and destroy unless hazardous chemicals, mechanical abrasion, or thermal treatments are employed. Many of these treatments are labor-intensive, expensive, or present environmental and health concerns. To address these issues, we have developed a novel silyl-based technology that imparts degradable capabilities to crosslinked polyurethane networks, such as coatings. These silyl-containing polyurethanes are thermally stable and highly crosslinked, yet they will disassemble on demand via cascading bond cleavage when exposed to a selective, mild, and environmentally friendly fluoride salt solution. Furthermore, these polyurethanes are resistant to disassembly with strong acid and base solutions, which demonstrates their selectivity and robustness compared to other degradable crosslinked networks.
Introduction
Crosslinked materials are formed by the heat- or radiation-initiated chemical reaction of molecules to generate a permanent three-dimensional (3D) network. Once formed, these materials are irreversible, cannot be solvated, and cannot be heated and reformed. Crosslinked networks are found in numerous high-performance materials, such coatings, composites, sealants, and adhesives. For coatings, the crosslinked network imparts unique properties, such as hydrocarbon resistance, increased thermal stability, and enhanced mechanical properties. As a result, these coatings are often used as topcoats on automobiles, aircraft, marine vessels, and bridges, or as anti-corrosive primers for pipelines, water tanks, and oil platforms. Although crosslinked coatings offer a variety of properties to enhance longevity and performance, they are simultaneously difficult to degrade and destroy because of their complex network of tangled polymeric chains and covalently bonded linkages.
Methods of Coating Degradation and Removal
Crosslinked coatings are currently degraded and removed from substrates using hazardous chemicals, abrasive media, or thermally ablative treatments, yet each of these methods has issues. For example, the primer and topcoat on U.S. Navy aircraft are often chemically stripped with methylene chloride or benzyl alcohol blends, which generate large quantities of hazardous waste and release substantial amounts of volatile organic compounds (VOCs) into the air. Methylene chloride strippers are the least expensive and the most effective method of chemically removing coatings because they rapidly swell, soften, and delaminate the crosslinked networks (Figure 1a).1 However, methylene chloride is considered to be extremely toxic, resulting in numerous health issues and even death due to asphyxiation.2 As a result, in March 2019, the U.S. Environmental Protection Agency (EPA) banned the sale of methylene chloride paint removers to consumers, although the agency continued to allow its use in commercial applications for the foreseeable future.3a,b Benzyl alcohol strippers and blends thereof are less hazardous than methylene chloride, but they are slower acting and require multiple treatments, resulting in increased waste, reduced operational readiness, and increased maintenance costs. Furthermore, the use of acidic benzyl alcohol strippers on DoD aircraft is prohibited because they have been shown to cause hydrogen embrittlement of high-strength steel.4
Stripping of coatings via abrasive blast media (e.g., steel shot, plastic aggregate) or mechanical sanders requires specialized equipment, environmental containment, and generates hazardous airborne particles (Figure 1b). Hand-sanding is less expensive, but it is time-consuming and labor-intensive. Laser ablation, which is a thermal treatment, generates minimal waste and has proven to be an efficient method for the de-painting of U.S. Air Force aircraft.5,6 However, lasers are expensive and require highly trained individuals or robotics, and coatings on complex geometries are difficult to remove. Although our examples focus on military aerospace coatings, there are numerous nonmilitary markets where these coating removal methods are utilized.

Chemically Degradable Networks
Commercial crosslinked coatings are not designed to be chemically stripped with a selective chemical, and there are no examples in the literature that describe a crosslinked coating being selectively degraded and removed from a substrate via a mild chemical treatment at room temperature. Academic research on the chemical degradation of crosslinked networks, such as coatings, has focused on developing materials that possess cleavable bonds and linkages.7 For instance, olefinic bonds in epoxy-amine networks were cleaved with potassium permanganate in acetic acid, acetal groups in anhydride-cured networks were cleaved by heating with a solution of phosphoric acid, and tertiary carbamate groups in epoxy-amine networks were cleaved with heated solutions of mineral acids.8,9 In 2014, poly(hexahydrotriazine) (PHT) networks formed from aromatic diamines and paraformaldehyde under thermal conditions were disassembled by reversing the diamines with a solution of sulfuric acid.10 In all of these cases, highly acidic (i.e., pH < 2) conditions, and often elevated temperatures, were required to initiate cleavage of the covalent bonds and linkages. Furthermore, none of these networks possess properties (e.g., flexibility, weatherability) that are typically found in commercial coatings, let alone properties that are necessary for high-performance applications.
Degradable Silyl-Containing Polyurethanes
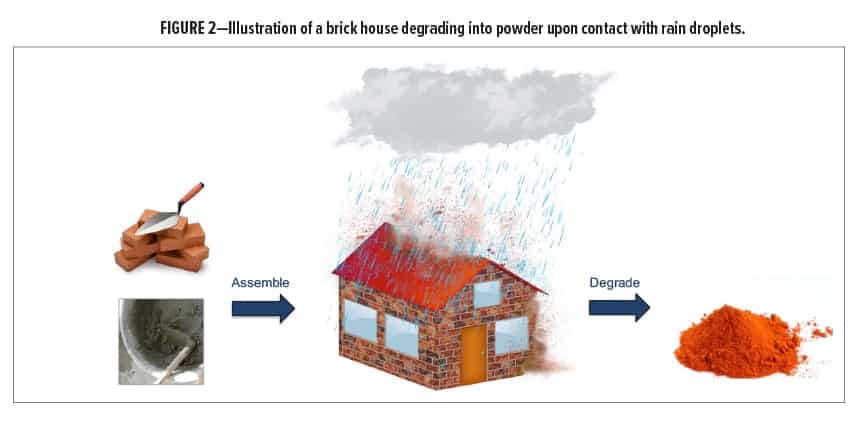
The U.S. Naval Research Laboratory has recently developed novel silyl-containing crosslinked materials that possess similar chemical linkages and polymeric chains as those found in commercial two-component (2K) polyurethane coatings. However, unlike commercial 2K polyurethanes, these silyl-containing networks degrade in multiple directions via cascading bond cleavage when treated with a specific chemical reagent. An increase in reaction entropy assists with facilitating network degradation, which results in the formation of smaller molecules than those used to form the networks.11,12 The concept is akin to a house that is built from bricks and cement yet degrades into powder when exposed to rain droplets (Figure 2). Herein, we discuss the synthesis, properties, and potential coating applications of crosslinked silyl-containing polyurethanes that can be selectively disassembled on demand with an environmentally friendly fluoride salt.
Experimental Procedures
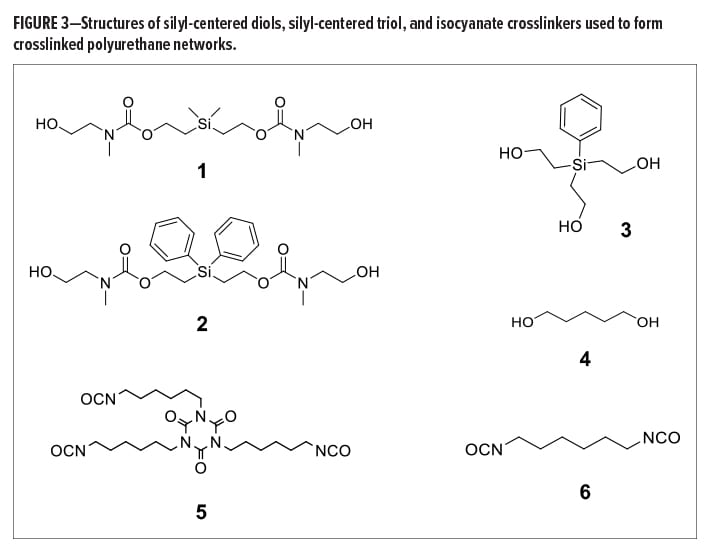
Aliphatic silyl-centered diols and triols were synthesized via multi-step procedures. Non-extended chain silyl-centered diols were synthesized from diphenyldivinylsilane or dimethyldivinylsilane via a hydroboration reaction with 9-Borabicyclo[3.3.1]nonane (9-BBN) in tetrahydrofuran (THF), followed by an oxidative work-up. Extended chain silyl-centered diols, such as (dimethylsilanediyl)bis(ethane-2,1-diyl) bis((2-hydroxyethyl)(methyl)carbamate) (1) (Figure 3) and (diphenylsilanediyl)bis(ethane-2,1-diyl) bis((2-hydroxyethyl)(methyl)carbamate) (2), were synthesized from the corresponding non-extended diols via a two-step process. This consisted of a carbonate intermediate that was formed from reaction with N,N’-disuccinimidyl carbonate (DSC) in acetonitrile, followed by carbamate formation using N-methylethanolamine in basic acetonitrile. These procedures are discussed in broader detail in the Supporting Information of reference (12). Silyl triol, 2,2’,2’’-(phenylsilanetriyl)tris(ethan-1-ol) (3), was synthesized via the hydro-boration of phenyltrivinylsilane using 9-BBN in THF, followed by oxidative work-up. Isocyanate-functional molecules, such as the isocyanurate trimer of hexamethylene-1,6-diisocyanate (HDI) (5) and monomeric HDI (6), were purchased from a commercial vendor and used as received. The structure of all synthesized molecules was confirmed via 1H and 13C nuclear magnetic resonance (NMR), Fourier transform infrared (FTIR) spectroscopy, and high-resolution mass spectrometry (HRMS).
Silyl-containing polyurethane networks were formed by dissolving aliphatic isocyanate 5 or 6 in THF in a round-bottom flask, followed by the addition of silyl diol or triol. The flask was then heated to 50°C and stirred for one hour, then the mixture was poured into a circular aluminum pan and heated overnight at 60°C in an oven. Silyl-containing polyurethanes based on 50/50 reactive equivalent blends of isocyanates 5 and 6 were also formed using this procedure. Non-silyl-containing polyurethanes based on 1,5-pentanediol (4) and these isocyanates were synthesized for use as controls. All networks were about 2 mm in thickness, which is substantially greater than the thickness of 2–3 mils (ca. 50–75 microns) for most commercial topcoats.
The thermal properties of the silyl-containing polyurethane networks were determined using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), whereas the surface and bulk chemistry were determined using attenuated total reflectance infrared (ATR-IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), and several other analytical instruments. The degree of crosslinking was determined by gel fraction calculations, whereas the degree of network swelling was determined using organic solvents and water.
Network disassembly was facilitated by immersing small pieces (i.e., 15 mm width x 20 mm length x 2 mm thick) of the silyl-containing polyurethane networks in a solution of tetrabutylammonium fluoride (TBAF) or cesium fluoride (CsF) in THF, acetone, or propylene glycol monomethyl ether acetate (PM Acetate) at room temperature. The solutions were either static (i.e., non-stirred) or dynamic (i.e., stirred with a magnetic stir bar). The degree of network disassembly was determined using ATR-IR, DSC, and confocal microscopy, whereas the small molecules formed during disassembly were determined using thermogravimetric analysis/mass spectrometry (TGA-MS) and gas chromatography/mass spectrometry (GC-MS).
Results
Synthesized silyl-centered molecules with varied compositions and electrophilicity demonstrated the ability to disassemble at different rates via cascading bond cleavage when exposed to fluoride ion,13 and we envisioned that functionalizing these molecules with terminal hydroxyl groups would enable the formation of novel crosslinked networks with selectively degradable capabilities.
Silyl-Containing Polyurethanes and Characterization
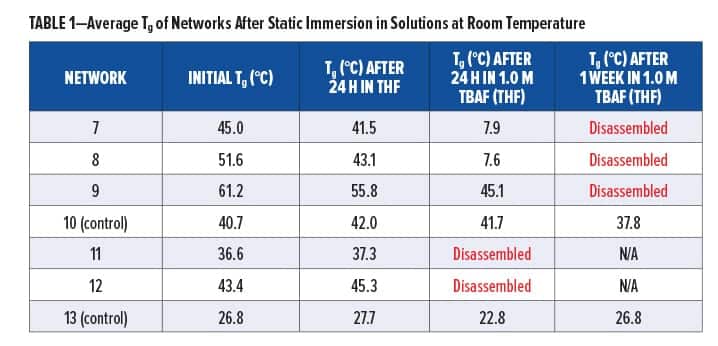
Synthesized silyl diols 1 and 2 were used to form silyl-containing polyurethane networks 7 and 8, respectively, whereas silyl triol 3 was used to form network 9 (Figure 4). The non-silyl-containing control (10) was formed from non-silyl diol 4. ATR-IR spectra showed that no isocyanate groups remained in the networks, and XPS confirmed the presence of silicon in polyurethanes 7, 8, and 9. The gel fraction of all materials was greater than 0.97, which is consistent for highly crosslinked networks. Silyl-containing polyurethanes 7 and 8 possessed glass transition temperatures (Tgs) of 45.0°C and 51.6°C, respectively, whereas network 9 had a Tg of 61.2°C and the control (10) had a Tg of 40.7°C. The higher Tg for polyurethane network 9 is likely due to the shorter aliphatic chains lengths between silicon and the carbamate linkages, including the three crosslinks per silyl molecule, whereas the control likely demonstrated the lowest Tg due to its largely aliphatic chain structure and lack of geminal methyl or phenyl groups between the carbamate linkages. All polyurethane networks had onset degradation temperatures above 286°C, which indicated good thermal stability.
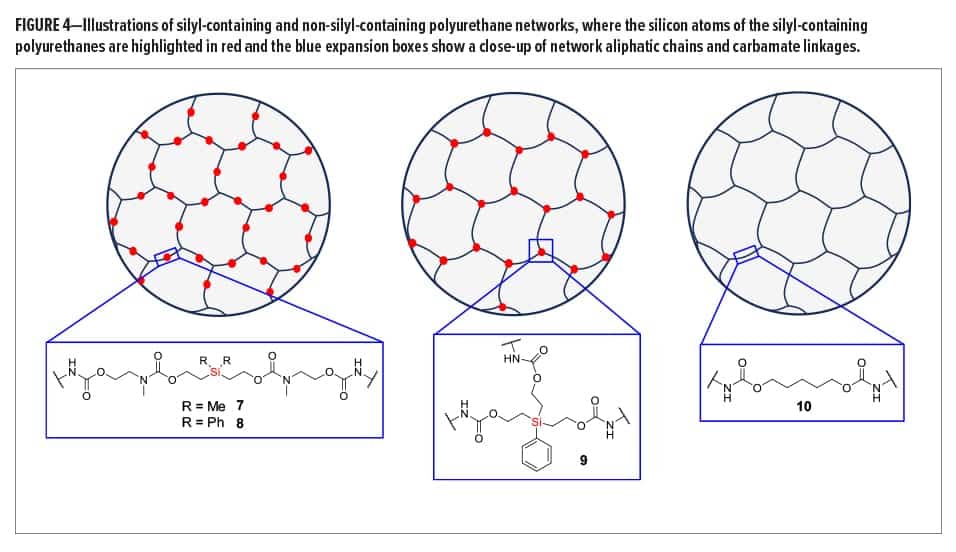
Silyl-containing polyurethanes with reduced crosslink density (11 and 12) were synthesized from silyl diol 1 or 2 and a 50/50 reactive equivalent mixture of isocyanates 5 and 6. A non-silyl-containing control (13) was also synthesized for comparison. Similar to networks 7, 8, and 9, these polyurethanes showed no remaining isocyanate groups when tested via ATR-IR, and the gel fraction of all networks was greater than 0.98. However, the decreased crosslink density for these networks resulted in reduced glass transition temperatures, as the Tgs for 11, 12, and 13 were 36.6°C, 43.4°C, and 26.8°C, respectively. Similar to the networks with higher crosslinked density, the control (13) possessed the lowest Tg, which is likely the result of greater bond rotation within the pentylene chains compared to the silyl-containing chains of networks 11 and 12. Furthermore, the phenyl groups of polyurethane 12 resulted in the highest Tg of the three because of reduced bond rotation within the chains.
Exposure of Silyl-Containing Polyurethanes to Fluoride Salts
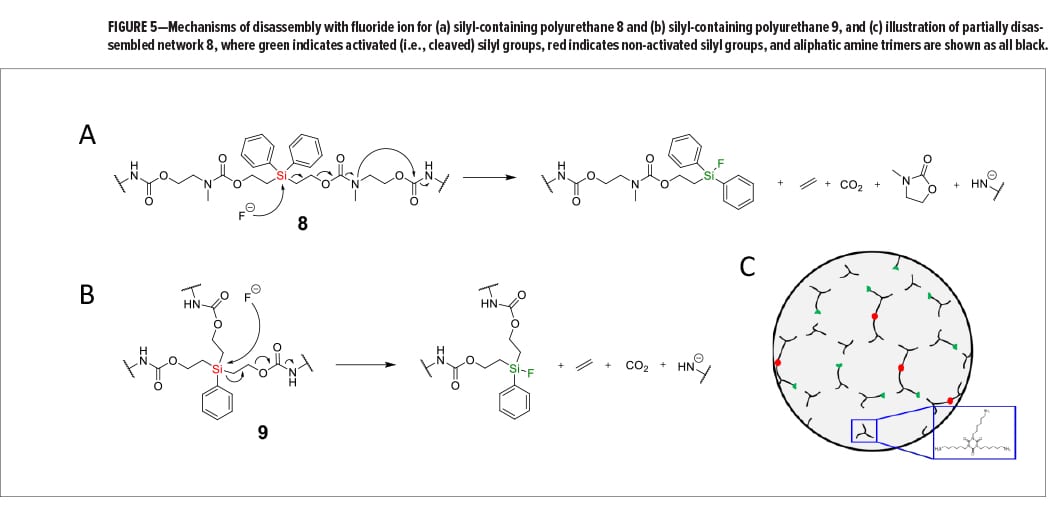
Fluoride salts were chosen as the selective chemical stimuli to disassemble the silyl-containing polyurethane networks because of their mild reactivity and environmentally friendly nature compared to methylene chloride, strong acids, strong bases, or other harsh chemicals. Fluoride salts were also chosen because of fluoride ion’s ability to form strong covalent bonds with silicon, in addition to fluoride ion’s inability to cleave linear aliphatic carbamate groups at room temperature.14 As shown in Figure 5a, we envisioned that reaction of fluoride ion with a silyl-containing chain in network 8 would cleave the Si–C bond and initiate a cascading breakage of bonds that would eliminate volatile ethylene and carbon dioxide, including cyclic molecule 3-methyloxazolidin-2-one, and result in the generation of an aliphatic amide ion. Not only would this reaction break several covalent bonds within the network, but it would also break the carbamate linkages generated during polyurethane network formation. For network 9, reaction with fluoride ion would cleave one of three Si–C bonds to eliminate both ethylene and carbon dioxide, resulting in an aliphatic amide ion (Figure 5b). Amide ions formed during disassembly would likely be protonated via water or another proton source to generate primary amines with hydrocarbon chains. Figure 5c illustrates network 8 when partially disassembled with fluoride ion, and where a resulting byproduct of disassembly is the aliphatic triamine 1,3,5-tris(6-aminohexyl)-1,3,5-triazinane-2,4,6-trione. Reaction of the remaining silyl groups with additional fluoride ion would, in theory, lead to disassembly of all remaining chains in the network.
Immersion of silyl-containing networks 7, 8, and 9 in static 1.0 M TBAF (THF) for 24 h at room temperature resulted in degradation for all materials, as indicated by their decreased Tg (Table 1). When analyzed with ATR-IR, we observed the formation of a new peak at 881 cm-1 in all networks, which is indicative of Si–F bond formation resulting from the reaction of fluoride ion with silyl groups. We also observed a decrease in the amide I peak at 1676 cm-1 and amide II peak at 1535 cm-1, as well as subsequent peak broadening in the NH peak at 3338 cm-1, which provided confirmation for loss of carbamate groups. The mechanisms of disassembly were confirmed via: (1) TGA-MS analysis, which showed the loss of ethylene and carbon dioxide, and (2) GC-MS analysis of extracts from network 8, which were found to contain cyclic molecule 3-methyloxazolidin-2-one. After 24 h, all networks were visually smaller in size and less rigid when handled. Disassembly was not observed in the polyurethane control (10).
Silyl-containing polyurethane 9 was found to completely disassemble after 36 h of static immersion in 1.0 M TBAF (THF), whereas five days of static immersion were required to completely disassemble network 8. As shown in Figure 6, network 8 expanded and broke into two pieces within three days of immersion in the fluoride salt solution, then visually disappeared on day five. For comparison, when network 8 was exposed to only THF for five days we observed zero disassembly. The faster disassembly for 9 is likely the result of its network possessing more cleavable bonds compared to 8, in addition to providing a larger entropic increase as disassembly occurs. Immersion of network 8 in a dynamic solution of 1.0 M TBAF (THF) led to complete disassembly in less than 24 h, which can be attributed to increased swelling of the network, increased collisions between fluoride ion and the silyl groups, and increased mobility of the disassembled molecules. As mentioned previously, these networks were about 2 mm thick, which is significantly thicker than most commercial topcoats. Thus, it is believed that thinner networks would disassemble significantly faster when exposure to fluoride salt solutions.

Alternative fluoride salts solutions, such as TBAF in acetone and cesium fluoride (CsF) in THF, have also shown the ability to disassemble these networks. For example, silyl-containing polyurethane 9 completely disassembled within three days of static immersion in 1.0 M TBAF (acetone), yet only partially disassembled upon static immersion in 0.5 M CsF (THF) after one week. The decrease in Tg for 9 was only 10.4°C after one week in CsF, which is probably due to the reduced concentration of fluoride ion and cesium fluoride’s reduced solubility in THF compared to TBAF. It is worth mentioning that no disassembly or change in chemical bonds for 8 and 9 was observed when immersed in static 1.0 M TBAF (aq.), 1.0 M HCl (aq.), and 1.0 M NaOH (aq.) solutions, thus demonstrating that these silyl-containing networks possess high chemical stability and robustness compared to previously reported degradable networks.
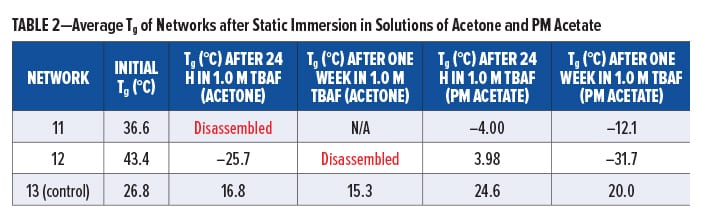
Silyl-containing polyurethane networks with reduced crosslink density (11 and 12), including the non-silyl-containing polyurethane control with reduced crosslink density (13), were exposed to static solutions of THF and 1.0 M TBAF in THF, acetone, and PM Acetate at room temperature. Disassembly of 11 and 12 did not occur in THF after 24 h, and their change in Tg was nominal (Table 1). A similar change in Tg was observed for the control (13). However, when networks 11 and 12 were immersed in 1.0 M TBAF (THF), they completely disassembled within 24 h (Table 1). This was significantly faster compared to networks 8 and 7, which required five days or more, respectively, to completely disassemble. Furthermore, as shown in Table 2, immersion of 11 and 12 in 1.0 M TBAF (acetone) resulted in disassembly within 24 h to one week, whereas immersion in 1.0 M TBAF (PM Acetate) only resulted in a decrease in Tg after one week and complete disassembly was not observed. The slower time for disassembly in PM Acetate compared to THF and acetone is likely the result of PM Acetate’s reduced ability to swell the polymeric chains of the silyl-containing polyurethanes. However, a longer time of exposure in 1.0 TBAF (PM Acetate), or use of dynamic conditions, would likely result in complete disassembly of 11 and 12. The purpose of evaluating acetone and PM Acetate was to determine if solvents more commonly found in paints and coatings could be used for disassembly.
Conclusions
In this article, we demonstrate that novel silyl-containing polyurethane networks can be synthesized and selectively degraded on demand, via cascading bond cleavage, using a fluoride salt solution at room temperature. The silyl-containing polyurethanes showed good thermal stability with degradation beginning around 290°C, which is typical for a crosslinked polymeric coating. Exposure of the silyl-containing polyurethanes to THF, strong acid, and strong base resulted in zero-to-minor changes in chemical structure and thermal properties, whereas changes were pronounced when exposed to fluoride salts in organic solvents. Silyl-containing polyurethanes with reduced crosslink density and glass transition temperature were found to degrade more rapidly upon exposure, which is likely the result of fluoride ion’s ability to more easily penetrate their networks as the outer polymeric chains are disassembled. This silyl-containing technology has potential applications as high-performance coatings that can be selectively degraded and removed from a substrate using an environmentally friendly fluoride salt stripper, rather than using hazardous chemical strippers or abrasive materials.
Acknowledgments
This research was supported by the U.S. Naval Research Laboratory’s Base Program.
References
1. Durkee, J., “Paint Stripping: It’s Just Like Parts Cleaning,” Met. Finish., 107, 49 (2009).
2. MacIsaac, J., Harrison, R., Krishnaswami, J., McNary, J., Suchard, J., Boysen-Osborn, M., Cierpich, H., Styles, L., and Shusterman, D., “Fatalities Due to Dichloromethane in Paint Strippers: A Continuing Problem,” Am. J. Ind. Med., 56, 907 (2013).
3. (a) “EPA Bans Consumer Sales of Methylene Chloride Paint Removers, Protecting Public,” https://www.epa.gov/newsreleases/epa-bans-consumer-sales-methylene-chloride-paint-removers-protecting-public, 15 March 2019, (b) “EPA’s Methylene Chloride Ban Excludes Workers,” https://www.webmd.com/lung/news/20190318/epa-methylene-chloride-ban-excludes-workers, 18 March 2019.
4. “Benzyl Alcohol Paint Stripping,” https://p2infohouse.org/ref/20/19926/P2_Opportunity_Handbook/5_9.html, August 2001.
5. Mongelli, G., “Portable Laser Coating Removal System (PLCRS),” ESTCP WP-200027, September 2005.
6. Hoehman, T., “Robotic Laser Coating Removal System (RLCRS),” ESTCP WP-200526, August 2008.
7. Ma, S. and Webster, D.C., “Degradable Thermosets Based on Labile Bonds or Linkages: A Review,” Prog. Polym. Sci., 76, 65 (2018).
8. Griffith, J.R., “Epoxy Resins Containing a Specific Vulnerability,” ACS Symp. Ser., 114, 259 (1979).
9. Buchwalter, S.L. and Kosbar. L.L., “Cleavable Epoxy Resins: Design for Disassembly of a Thermoset,” J. Polym. Sci., Part A: Polym. Chem., 34, 249 (1996).
10. Garcia, J.M., Jones, G.O., Virwani, K., McCloskey, B.D., Boday, D.J., ter Huurne, G.M., Horn, H.W., Coady, D.J., Bintaleb, A.M., Alabdulrahman, A.M.S., Alsewailem, F., Almegren, H.A.A., and Hedrick, J.L., “Recyclable, Strong Thermosets and Organogels via Paraformaldehyde Condensation with Diamines,” Science, 344, 732 (2014).
11. Iezzi, E.B., Camerino, E., Daniels, G., and Wynne, J.H., “Silyl-Containing Alcohols and Amines for Thermosets that Disassemble On-Demand,” U.S. Patent Appl. No. 15/843,181, 15 December 2017.
12.Daniels, G.C., Camerino, E., Wynne, J.H., and Iezzi, E.B., “Cross-Linked Networks that Selectively and Controllably Disassemble On-Demand via Cascading Bond Cleavage,” Mater. Horiz., 5, 831 (2018).
13. Camerino, E., Daniels, G.C., Wynne, J.H., and Iezzi, E.B., “Synthesis and Kinetics of Disassembly for Silyl-Containing Ethoxycarbonyls using Fluoride Ions,” RSC Adv., 8, 1884 (2018).
14. Jacquemard, U., Bénéteau, V., Lefoix, M., Routier,S., Mérour, J.-Y., and Coudert, G., “Mild and Selective Deprotection of Carbamates with Bu4NF,” Tetrahedron, 60, 10039 (2004).
CoatingsTech | Vol. 17, No. 1 | January 2020